Installing DFTB in ASE
For instalation refer to this document or ASE documentation.
Solvation using DFTB
Making solvated layer with PACKMOL
For packmol instalation and general tutorial refer to PACKMOL website.
For further text it is assumed that packmol is properly instaled and can be called as $ packmol.
Adding water molecules to surface
The general packmol input code looks like this
seed GEN_SEED
tolerance 2.0
output GEN_OUTPUT
filetype xyz
structure GEN_OUTPUT
constrain_rotation x 0. 0.
constrain_rotation y 0. 0.
constrain_rotation z 0. 0.
number 1
inside box 0. 0. 0. 12.873228257488138 12.368764968332382 5
end structure
structure h2o.xyz
number GEN_WATER_COUNT
inside box 1. 1. 1. 11.873228257488138 11.368764968332382 5.
end structure
To explain the code:
- seed is an integer to initialise randomness, use -1 for current time based seed, or any integer for reproducable seed
- tolerance parameter describes minimal distance between packed molecules in AA.
- first
structure GEN_INPUTblock puts the surface at the bottom of box, z height is chosen to be 5, as it must accomodate the height of adsorbed molecule.
Box size is chosen to match the cell size for the surface - Next
structure h2o.xyzblock puts GEN_WATER_COUNT molecules (replace with desired integer) in the box, using predefined water molecule in fileh2o.xyz. For close layer of water molecules on the surface z = 5 is good, but can be changed as desired.
Note the reduced box cell size, since PACKMOL does not support periodic boundary conditions, and given full cell would place molecules in such a way that there are clashes along periodic boundaries.
Helper utility for easier use
We can use CLI utility SED so that we can create a single base input file and easily apply to multiple surfaces without manualy editing the code.
Additionaly with this we can set periodic cell, since packmol script deletes previous cell information.
#!/bin/bash
# Set parameters using command line arguments
# example call:
# $ sh solvate_slab.sh co_surf_o_nosolv_opt.xyz co_surf_o_50H2O.xyz 50 -1
input_filename=$1
output_filename=$2
number_of_water_mol=$3
seed=$4
# Generate packmol input file using passed arguments
cp generic_packmol_input.inp packmol.inp
sed -i "s/GEN_SEED/$seed/" packmol.inp
sed -i "s/GEN_OUTPUT/$output_filename/" packmol.inp
sed -i "s/GEN_INPUT/$input_filename/" packmol.inp
sed -i "s/GEN_WATER_COUNT/$number_of_water_mol/" packmol.inp
# Run packmol using generated input, then add proper cell parameters for ASE
packmol < packmol.inp
lattice_string='Lattice="12.873228257488138 0.0 0.0 0.0 12.368764968332382 -4.481704022984947e-13 0.0 -3.9543094456634876e-13 14.000000000000002" Properties=species:S:1:pos:R:3'
sed -i "2s/.*/$lattice_string/" $output_filename
Now we can copy general packmol code, predefined water molecule file and helper bash script in the folder of surface molecule and run it as:
$ sh solvate_slab.sh co_surf_o_nosolv_opt.xyz co_surf_o_50H2O.xyz 50 -1
to view the obtained structure we can use either vmd or ase gui
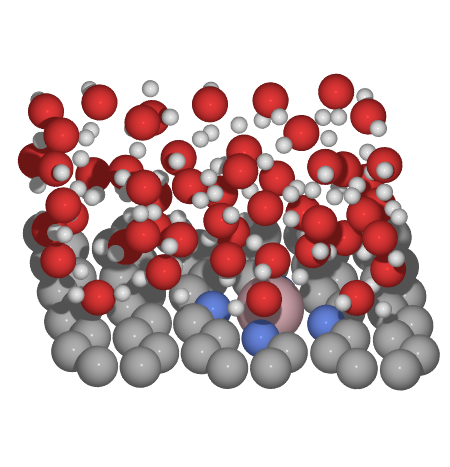
Using DFTB to optimize obtained solvated surface
To optimize obtained solvated surface we can use following python code (assuming DFTB is set up properly).
# Python script to optimize molecule using DFTB
# example usage:
# $ python dftb_opt_solvated.py co_surf_o_50H2O.xyz 1
from ase.constraints import FixAtoms
from ase.calculators.dftb import Dftb
from ase.optimize import QuasiNewton
from ase.io import read, write
import sys
# Read structure name from command line
fname=sys.argv[1]
surface_atoms=int(sys.argv[2])
solvated_molecule = read(fname)
# Constrain surface, so that only water layer is optimized
c = FixAtoms(indices=range(0, 59+surface_atoms))
solvated_molecule.set_constraint(c)
# Set DFTB parameters
calc = Dftb(atoms=solvated_molecule,
label='solvated_opt',
Hamiltonian_MaxAngularMomentum_='',
Hamiltonian_MaxAngularMomentum_O='p',
Hamiltonian_MaxAngularMomentum_N='p',
Hamiltonian_MaxAngularMomentum_C='p',
Hamiltonian_MaxAngularMomentum_Co='d', # Change if not Cobalt
Hamiltonian_MaxAngularMomentum_H='s',
Options_WriteDetailedXml = 'Yes',
kpts=(1,1,1)
)
solvated_molecule.calc = calc
solvated_molecule.get_potential_energy()
# Set optimization to force convergence less than 0.01eV/AA
dyn = QuasiNewton(solvated_molecule, trajectory='solvation_opt.traj')
dyn.run(fmax=0.01)
# Save optimized structure with opt_* prefix
write("opt_"+fname, solvated_molecule)
with this we optimize solvated slab using
$ python dftb_opt_solvated.py co_surf_o_50H2O.xyz 1
after about 6 minutes we obtain optimized structure with forces in system less than 0.01eV/AA
...
BFGSLineSearch: 552[765] 02:38:47 -8536.422948 0.0084
and we can visualise both, optimization trajectory from solvated_opt.traj and final structure from opt_co_surf_o_50H2O.xyz.
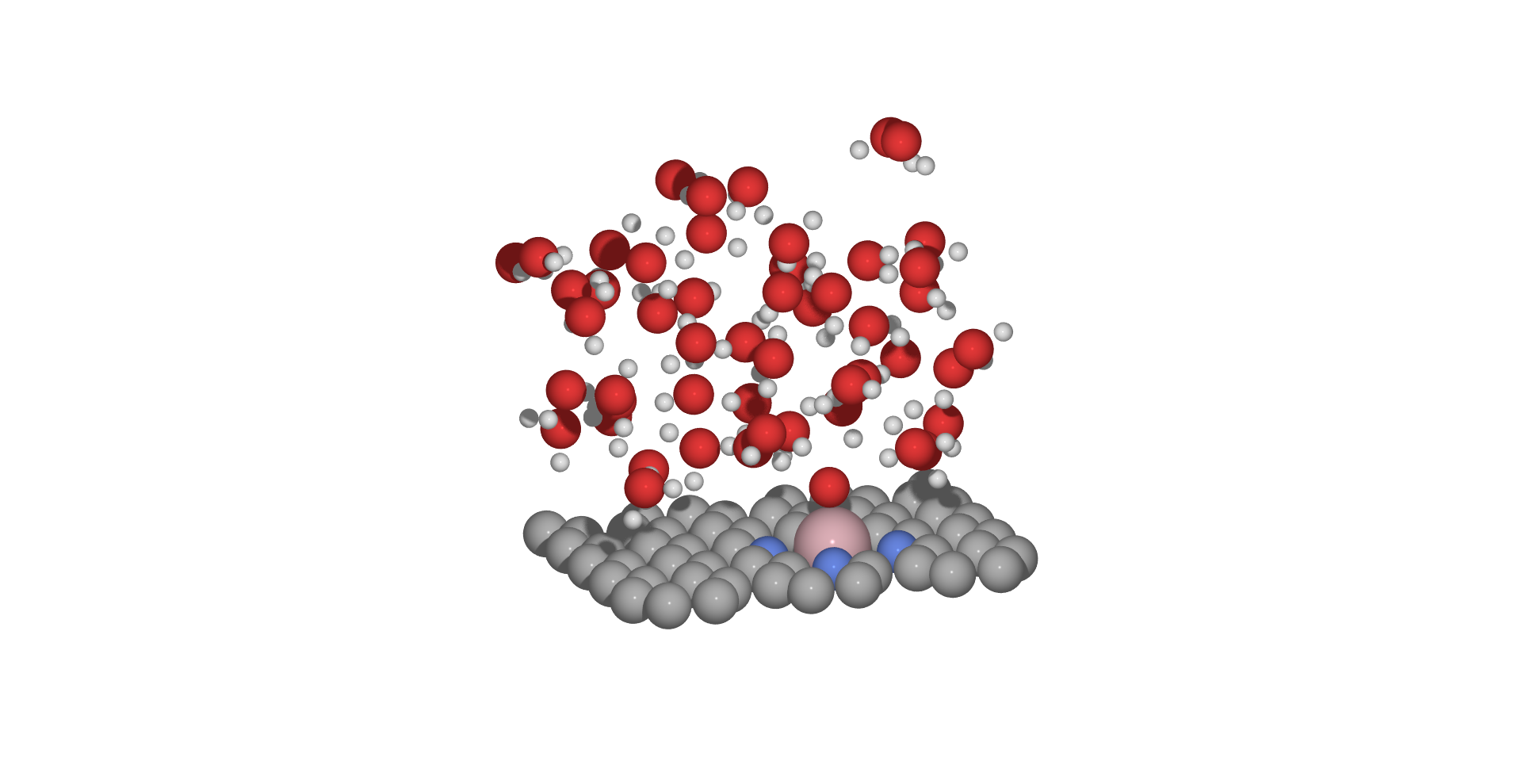
rerunning the procedure for different intial seed we obtain following result from optimization
BFGSLineSearch: 568[780] 02:56:59 -8536.439645 0.0098
at DFTB level the effect of different initial packing seems to be about 0.01eV which could be torelabe, resulting optimized structure single point DFT calculations to be done.