Working with vibrational analysis
More specificaly, vibrational analysis for adsorbates on surfaces
Small molecules
To calculate vibrational corrections for gas-phase molecules we require three parameters: symmetry number, geometry and spin.
For the symmetry number refer to the following table:

For geometry there are two options: linear or nonlinear
Spin should be obvious from the system.
from ase.vibrations import Vibrations
from ase.thermochemistry import IdealGasThermo
# Set up vibration calculations using previously optimized molecule
vib = Vibrations(molecule)
vib.run()
print(vib.summary())
vib_energies = vib.get_energies()
# Save vibration modes to review if imaginary frequency present
for mode in range( 3*len(molecule.numbers) ):
vib.write_mode(mode)
potentialenergy = molecule.get_potential_energy()
# Perform ideal gas calculations assuming ideal gas
thermo = IdealGasThermo(vib_energies = vib_energies,
potentialenergy = potentialenergy,
atoms=molecule,
geometry='nonlinear', # adjust this
symmetrynumber=2, # adjust this
spin=0 # adjust this
)
# Calculate respective corrections at temperature and pressure specified
G = thermo.get_gibbs_energy(temperature=298.15, pressure=101325.)
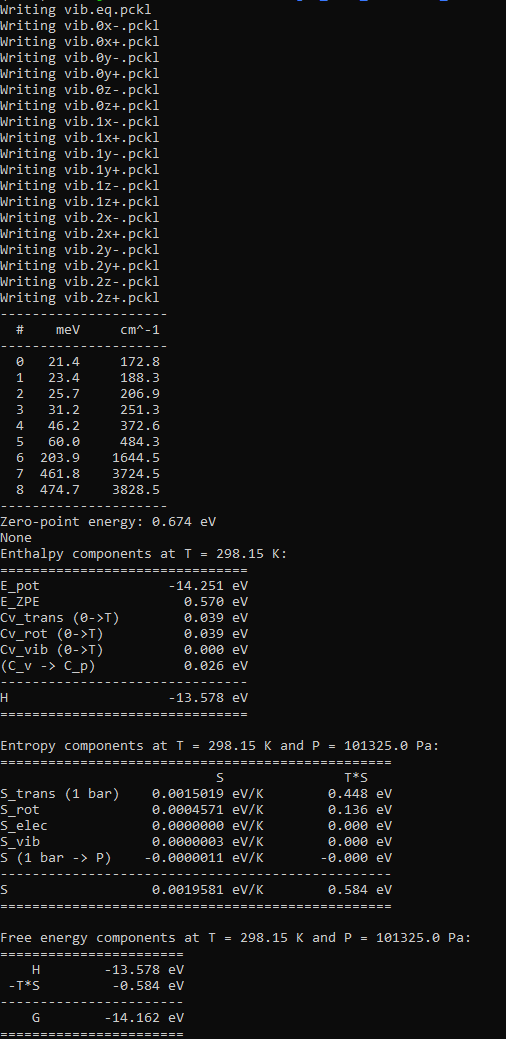
Example output for water:
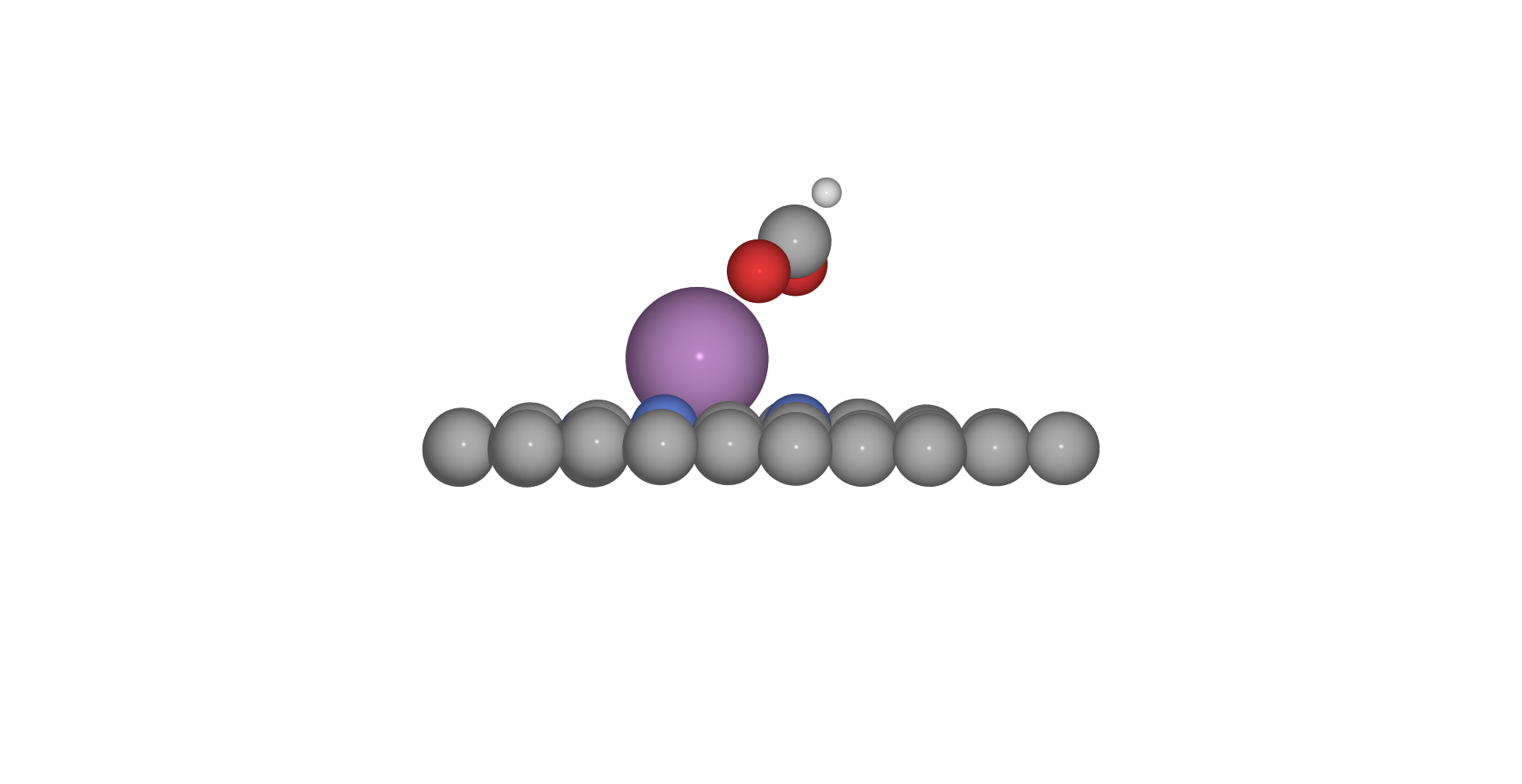
Surface adsorbates
To calculate vibrational corrections for adosrbates we use harmonic approximation and only consider the adsorbate molecule's vibrations. Here we fix the bulk slab and only vibrate the adsorbate atoms, which we assume in molecule are at the end. As such, we only need two parameters, the ID of last slab/surface atom and numer of atoms in adsorbate.
from ase.vibrations import Vibrations
from ase.thermochemistry import HarmonicThermo
# Sample parameters
last_slab = 58
No_vib_atoms = 3
# Vibrational calculation setup using previously optimized molecule
vib = Vibrations(molecule, indices = range(last_slab, last_slab+No_vib_atoms))
vib.run()
vib.summary()
# Save vibration modes to review if imaginary frequency present
for mode in range(3*No_vib_atoms):
vib.write_mode(mode)
# Print frequencies of vibrations
vib_energies = vib.get_energies()
print(vib_energies)
thermo = HarmonicThermo(vib_energies = vib_energies)
# Get thermodynamical parameters at required temperature
G = thermo.get_entropy(temperature=298.15)
G2= thermo.get_internal_energy(temperature=298.15)